I am a professor of Computer Science at MIT in the area of Computational Biology. I am a member of CSAIL and the Broad Institute of MIT and Harvard.
My research interests are in the area of computational biology, genomics, epigenomics, gene regulation, and genome evolution. Specifically:
in the area of genome interpretation, we seek to develop comparative genomics methods to identify genes and regulatory elements systematically in the human genome
in the area of gene regulation, we seek to understand the regulatory motifs involved in cell type specification during development, understand their combinatorial relationships, and how these establish expression domains in the developing embryo.
in the area of epigenomics, we seek to understand the chromatin signatures associated with distinct activity states, the changing chromatin states across different cell types and during differentiation, and the sequencing signals responsible for the establishment and maintenance of chromatin marks.
in the area of evolutionary genomics, understanding the dynamics of gene phylogenies across complete genes, the emergence of new gene functions by duplication and mutation, and the algorithmic principles behind phylogenomics.
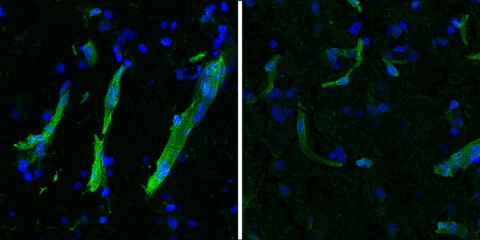
Study of 3.5 million cells from more than 100 human brains finds Alzheimer’s progression — and resilience to disease — depends on preserving epigenomic stability.
Genomics and lab studies reveal numerous findings, including a key role for Reelin amid neuronal vulnerability, and for choline and antioxidants in sustaining cognition.
MIT CSAIL and University of Tokyo researchers present GenoBoost, a polygenic scoring approach that accounts for both additive and non-additive genetic dominance effects in individuals’ health data.
Single-cell gene expression patterns in the brain, and evidence from follow-up experiments, reveal many shared cellular and molecular similarities that could be targeted for potential treatment.
Researchers looked at how non-coding regulatory regions are regulated in the four tissues of brain, heart, muscle, and lung, illuminating the intricate links between genetics, epigenetics, and disease mechanisms in a step towards more personalized medicine.





{kind=link}